


Regulatory Landscape and Approval Pathways for Active Implantable Medical Devices

The active implantable medical devices market operates within one of the most highly regulated environments in the healthcare industry. Because these devices are implanted in the human body and often support vital physiological functions, regulatory authorities impose strict requirements on safety, performance, and clinical effectiveness.
Understanding the regulatory landscape is essential for manufacturers, healthcare providers, and investors. Regulatory approval processes significantly influence product development timelines, market entry strategies, and overall competitiveness.
Regulatory Bodies and Frameworks
Key regulatory authorities include the U.S. Food and Drug Administration (FDA), the European Medicines Agency and Notified Bodies under the EU Medical Device Regulation (MDR), and regulatory agencies in Asia-Pacific such as Japan’s PMDA and China’s NMPA.
These organizations evaluate implantable devices based on clinical evidence, risk classification, and manufacturing quality standards. Active implantable medical devices are typically classified as high-risk products, requiring rigorous premarket evaluation.
Clinical Trials and Evidence Requirements
Manufacturers must conduct extensive clinical trials to demonstrate device safety and effectiveness. These trials often involve long follow-up periods to assess long-term performance and potential complications.
The need for high-quality clinical evidence increases development costs and extends time to market. However, it also ensures that only safe and effective devices reach patients.
Impact of EU MDR and Global Harmonization
The implementation of the EU MDR has significantly increased regulatory requirements for implantable devices in Europe. Stricter clinical evidence, post-market surveillance, and traceability rules have increased compliance costs for manufacturers.
At the same time, global efforts toward regulatory harmonization are helping streamline approval processes in some regions, enabling faster access to international markets.
Post-Market Surveillance and Reporting
Post-market surveillance is a critical aspect of implantable device regulation. Manufacturers are required to monitor device performance, report adverse events, and implement corrective actions when necessary.
These requirements improve patient safety and support continuous product improvement.
Conclusion
Regulation is a defining factor in the active implantable medical devices market. While regulatory requirements increase complexity and cost, they also ensure high standards of patient safety and product quality, supporting long-term market credibility.
Related Reports @
Implantable Medical Devices Market Size and Competitive Analysis by 2030
Wearable And Implantable Medical Devices Market Size, Share, Growth and Trends by 2034
Nanotechnology in Medical Devices Market Strategies, Top Players, Growth Opportunities, Analysis and Forecast by 2031
About Us -
The Insight Partners provides comprehensive syndicated and tailored market research services in the healthcare, technology, and industrial domains. Renowned for delivering strategic intelligence and practical insights, the firm empowers businesses to remain competitive in ever-evolving global markets.
Contact Information
Email: sales@theinsightpartners.com
Website: theinsightpartners.com
Phone: +1-646-491-9876
Also Available in : Korean German Japanese French Chinese Italian Spanish
Categorias
Leia Mais
The Shift Toward Private Stays Feels Real Now Something changed in the way people travel. Not overnight, but you can feel it. Hotels still exist, sure, but more travelers are quietly moving toward spaces that feel… theirs. A st barth villa rental isn’t just about a place to sleep. It’s about control. Your schedule, your space, your noise level. No elevator waits. No...

Basketball is not just a sport; it’s a passion that brings people together for intense competition and endless fun. If you’re someone who enjoys the thrill of scoring the perfect shot or pulling off impressive dribbles, then you might want to dive into the exciting world of Basketball Stars. This fast-paced game combines the essence of traditional basketball with unique...

Resource Utilization Matrix & Excel for Payroll.Master resource utilization matrix and excel for payroll to track efficiency, manage salaries, and boost business productivity easily! Resource Utilization Matrix & Excel for Payroll Made Easy Running a business smoothly is all about smart management—and that’s where a resource utilization matrix and excel for payroll come into...
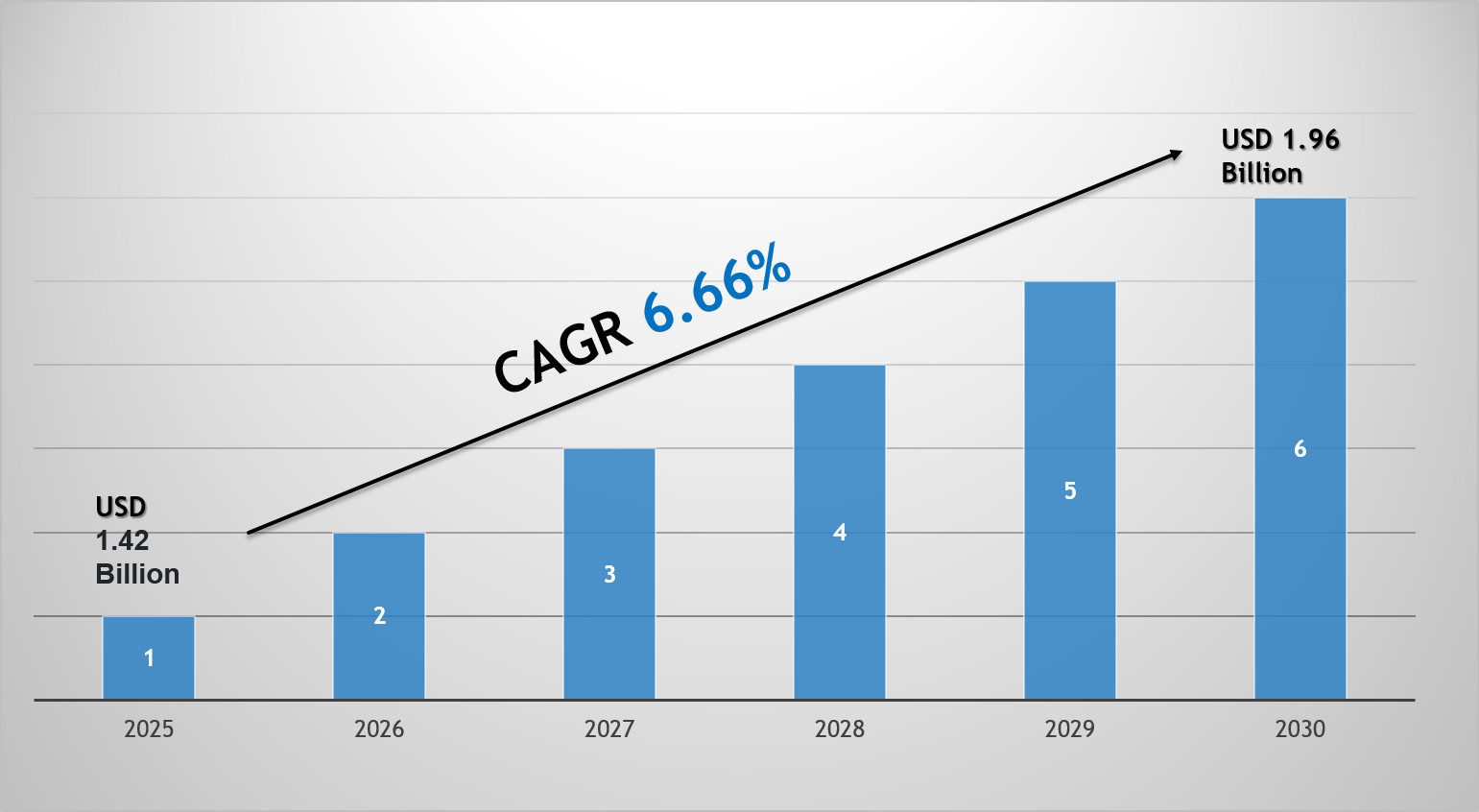
Future Medical Oxygen Concentrator Market: Key Dynamics, Size & Share Analysis The Global Medical Oxygen Concentrator Market size is valued at around USD 1.42 billion in 2025 and is projected to reach USD 1.96 billion by 2030. Along with this, the market is estimated to grow at a CAGR of around 6.66% during the forecast period, i.e., 2025-30. The market growth is primarily driven...
High Oleic Oil Market Summary “The global High Oleic Oil Market is expected to reach to USD 8.5 billion by 2032, growing at a CAGR of 9.4% during 2025 to 2032” The TrendBridge Insights Research report, “Global High Oleic Oil Market Report 2025 – Future Opportunities, Latest Trends, In-depth Analysis, and Forecast To 2032”, delivers strategic insights into...